Son un grupo diverso de enfermedades metabólicas, generalmente heredadas, ocasionadas por la ausencia de las enzimas que intervienen en la síntesis (producción) de la hemoglobina, sustancia necesaria para el transporte de oxígeno y parte esencial de los glóbulos rojos.
Cuando estas enzimas no están presentes, algunas sustancias que no pueden ser bien procesadas, se acumulan en los tejidos, estas sustancias que se acumulan son llamadas porfirinas.
Existen al menos 8 tipos de porfirias y por lo tanto existen varias clasificaciones. Según el sitio donde falte la enzima, las porfiras se dividen en: porfirias eritropoyéticas (médula osea) y hepáticas (higado); sin embargo desde el punto de vista dermatológico tenemos las porfirias con manifestaciones cutáneas y las que no las tienen. Otra clasificación es según los síntomas, podemos hablar de las porfirias agudas y las no agudas.
Las porfirias involucran tres síntomas principales:
- Cólicos o dolor abdominal (algunas formas de la enfermedad).
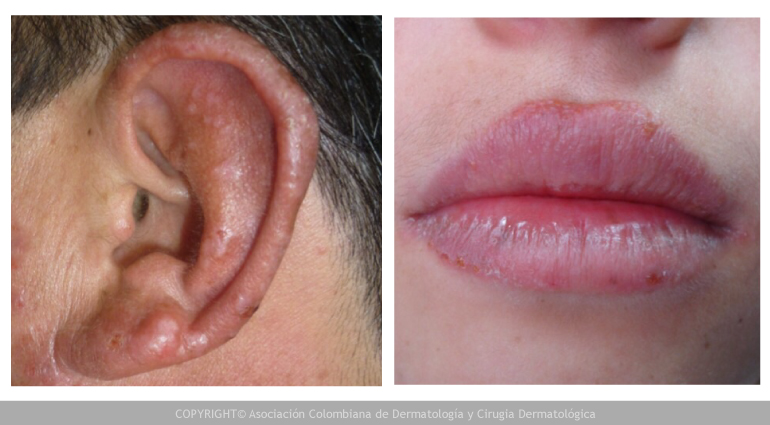
- Sensibilidad a la luz que causa erupciones y cicatrización de la piel (fotodermatitis).
- Problemas con el sistema nervioso (convulsiones, alteraciones mentales).
Los ataques pueden ocurrir en forma súbita, generalmente con dolor de estómago fuerte, seguido de vómito y estreñimiento. La exposición al sol puede causar dolor, sensaciones de calor, ampollas, al igual que enrojecimiento e hinchazón de la piel. Las ampollas sanan lentamente, a menudo con cicatrización o cambios en el color de la piel, y pueden ser deformantes. La orina se puede tornar de color rojo o marrón después de un ataque.
Otros síntomas son: Dolor muscular, parálisis o debilidad muscular, entumecimiento u hormigueo, dolor en brazos y piernas, dolor de espalda y cambios de personalidad. Los ataques agudos algunas veces son potencialmente mortales.
Para el diagnóstico se hacen necesarias las pruebas de laboratorio especiales. También se pueden realizar pruebas genéticas para detectar las mutaciones en los genes que producen las enzimas deficientes.
La prevención es extremadamente importante. En las porfirias agudas, se deben conocer y evitar los factores precipitantes de estas crisis; entre los principales están: el ayuno, alcohol, ciertos medicamentos, anestesias, hormonas, infecciones, estrés y tabaco.
En las porfirias que cursan con afectación de piel, evitar la exposición solar es lo más importante, ya sea con la no exposición, o con la aplicación de filtros o pantallas solares o el uso de prendas con filtro.
El tratamiento de la porfiria aguda es en general intrahospitalaria y de manejo exclusivo del personal médico y para las porfirias no agudas, evitar los factores desencadenantes y la fotoprotección.
Artículo escrito por:
Dra. María Claudia Guzmán S. MD
Residente Dermatología Universidad Autónoma de Bucaramanga.
Miembro Aspirante AsoColDerma